 Hotline服務熱線:010-61006450
Hotline服務熱線:010-61006450
 簡體中文
簡體中文體內外相關性在仿制藥中應用案例
1.背景
固體制劑開發過程中,溶出曲線常被用來評估藥品的生產工藝和產品質量。溶出曲線是指,在單位時間內,藥物在不同pH介質(模擬人體消化道的pH值)中的釋放曲線,常見溶出方法為籃法和槳法[1]。
檢測溶出曲線的另外重要目的是,評估藥物在體內的釋放情況。如何準確的評估藥物在體內釋放,可通過建立體內外相關性(IVIVR/C)模型,即利用藥品的溶出曲線,預測體內吸收。體內外相關模型的建立,能夠指導人體生物等效性研究,減少人體試驗次數,提高生物等效性試驗成功率,加快藥物的研發進度。
如何建立體內外相關性模型,我們首先要了解藥物在體內的轉運情況,即藥代動力學。藥代動力學包括藥物吸收、分布、代謝和消除。仿制藥開發過程中,人體生物等效性研究,作為判斷是否與原研藥質量和療效相當的一個金標準。
生物等效性研究一般在患者或健康受試者中進行,受試者服用藥物后,不同時間點采集血樣,檢測血液中藥物濃度,獲得血藥濃度—時間曲線。
2.不同制劑藥動學研究
為更好了解藥物的在體內的藥動學,下面會舉例說明溶液劑和固體制劑在體內吸收的異同。
溶液劑的藥代動力學:
受試者服用溶液劑后,藥物在體內沒有釋放過程,會被快速吸收,并分布及代謝和消除。所獲得的血藥濃度-時間曲線,如圖1所示。溶液劑不涉及藥物溶出過程,在體內的吸收系數、分布體積、消除系數均與原料藥本身的藥動學參數相關,藥物在體內的濃度可用指數方程C= C0e-kt來描述。
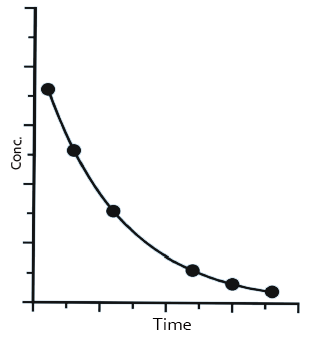
圖1—液體制劑血液濃度-時間曲線
固體制劑的藥代動力學:
當受試者服用固體制劑后,藥物首先在胃腸道中溶解,形成溶液后再吸收,然后分布,代謝及消除。所獲得的血藥濃度—時間曲線,如圖2所示。固體和液體制劑的藥動學曲線不同,是由于服用固體制劑后,藥物在體內會有緩慢釋放過程,而液體制劑沒有釋放過程。
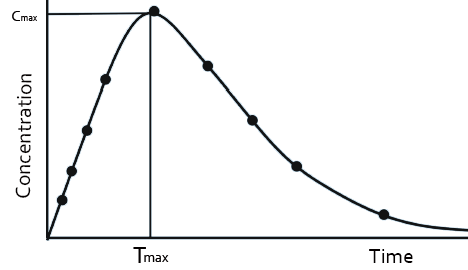
圖2 固體制劑-血藥濃度-時間曲線
由溶液制劑的藥動學曲線可知,藥物形成溶液后,在體內的轉運,僅與原料藥自身的藥動學參數有關。固體制劑的藥動學曲線,則與藥物在體內的溶解過程和藥物的本身屬性相關。而藥物在體內的溶解過程,可以用藥物的溶出曲線來表示,這也是體內外相關性模型建立的基礎。
一般來說,藥品的溶出不會改變藥物的藥代動力學,即藥物的藥動學參數,不會由于溶出的變化而變化。但不同的藥品的溶出曲線,能影響藥物在體內的溶解釋放速度,從而影響血液濃度—時間曲線。由此可知,藥代動力學為藥物的基本屬性,溶出則是藥品劑型的屬性。
3.IVIVR模型
體內外相關性模型,用IVIVR(in-vivo and in-vivo relationship)來表示,即將藥品的體外特性(主要為溶出曲線)和體內特性(藥動學)建立相關性。根據體外溶出數據,預測藥物的在體內的藥動學曲線,該方法為卷積法。相反,利用藥物的吸收曲線,可預測藥物在體內的溶出,該方法為反卷積法,如下圖所示:
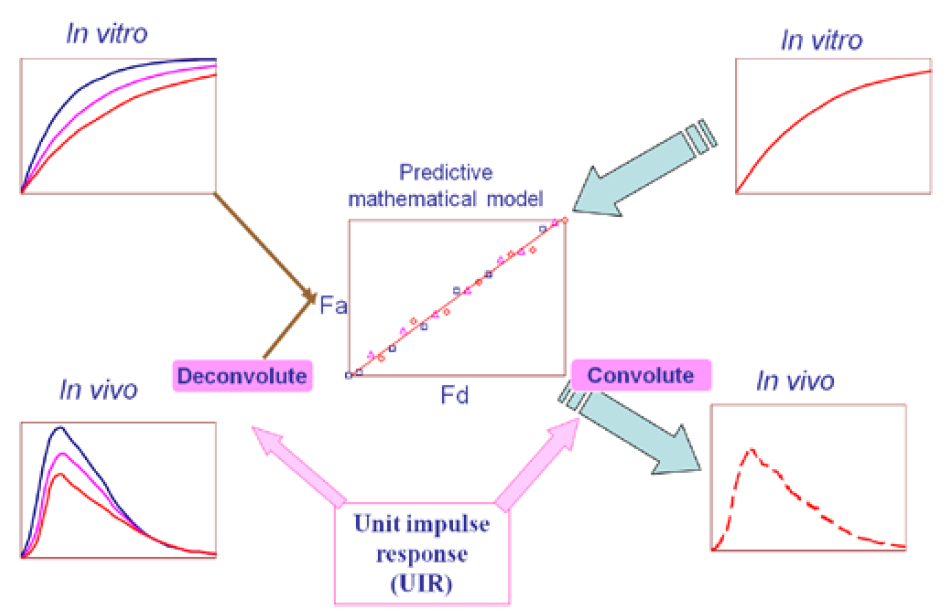
圖3 IVIVR建模過程-卷積與去卷積法示
IVIVR模型類型較多,常用體內外模型[2]的公式為:
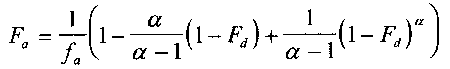
Fa:t時刻,藥物吸收的總量,
fa : t 時刻,藥物吸收的百分比,
α:一級滲透率常數(kpaap)與一級溶解率常數(kd)
Fd: t 時刻,藥物溶解的百分數。
IVIVR模型在仿制藥開發過程中,已有廣泛的應用,文獻報道[3,4],美托洛爾、吡羅昔康和雷尼替丁等項目,已成功利用該技術,進行處方工藝的開發。國內也應用IVIVR模型技術[5],建立不同劑型模型,實現了膠囊劑和分散片的生物等效性。
4.IVIVR應用案例
我司利用IVIVR模型,成功開發了鹽酸曲美他嗪片。曲美他嗪片臨床適用癥為抗心絞痛藥物,主要抑制游離脂肪酸代謝物,提高氧利用度,已緩解心肌缺血癥狀。體內外相關性模型中,輸入的藥物理化性質和藥動學參數如下:
藥物理化性質:
a. 曲美他嗪BCS III類,極易溶于水
b. pKa 4.45和 9.14 ,log P 1.04。
c. 溶出曲線:
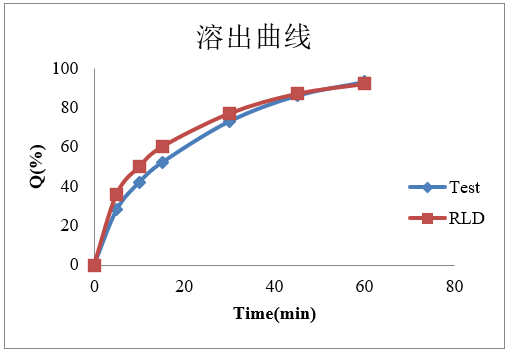
圖4-曲美他嗪片溶出曲線
藥動學性質:
a. 口服后,快速吸收,達峰時間少于兩小時。
b. 單次口服20毫克,達峰濃度約為55ng/ml
c. 連續給藥在24至36小時達到穩態,體內穩定性好。
d. 表觀分布容積為4.8 L/ kg,具有良好的組織分布特性
e. 蛋白結合率低:其體外測量值為16%。
f. 消除主要在尿液中,主要以原型藥形態,平均消除半衰期為6小時
參考文獻[6]“In vitro dissolution and in vivo bioequivalence evaluation of two brands of trimetazidine tables”,鹽酸曲美他嗪片的藥動學曲線如下:
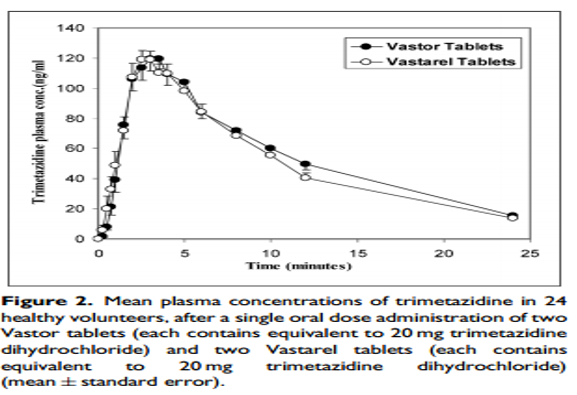
圖5 曲美他嗪PK曲線
嘗試將曲美他嗪體外溶出行為和體內吸收建立相關性,利用卷積計算方法,預測藥物在體內吸收。并將預測結果與文獻數據進行對比,判斷預測的準確性。文獻值與預測值對比:
|
Time |
文獻值 Vastarel Cp(ng/ml) |
Time |
預測值 R(pred) Cp(ng/ml) |
|
0.00 |
0.00 |
0.00 |
0.00 |
|
0.18 |
5.86 |
0.08 |
46.95 |
|
0.41 |
19.52 |
0.17 |
66.39 |
|
0.68 |
32.70 |
0.25 |
80.03 |
|
0.82 |
48.80 |
0.50 |
101.83 |
|
1.41 |
72.22 |
0.75 |
116.00 |
|
1.87 |
107.84 |
1.00 |
121.34 |
|
2.33 |
120.05 |
1.50 |
124.62 |
|
2.83 |
119.57 |
2.00 |
118.00 |
|
3.43 |
110.80 |
3.00 |
105.80 |
|
3.94 |
109.83 |
4.00 |
94.86 |
|
4.91 |
98.63 |
5.00 |
85.05 |
|
5.88 |
84.51 |
6.00 |
76.25 |
|
8.00 |
68.45 |
7.00 |
68.37 |
|
9.89 |
55.80 |
8.00 |
61.30 |
|
12.01 |
40.23 |
9.00 |
54.96 |
|
24.03 |
14.13 |
10.00 |
49.28 |
|
|
|
11.00 |
44.18 |
|
|
|
12.00 |
39.61 |
|
|
|
16.00 |
25.60 |
|
|
|
20.00 |
16.54 |
|
|
|
24.00 |
10.69 |
文獻值和預測值的PK曲線:
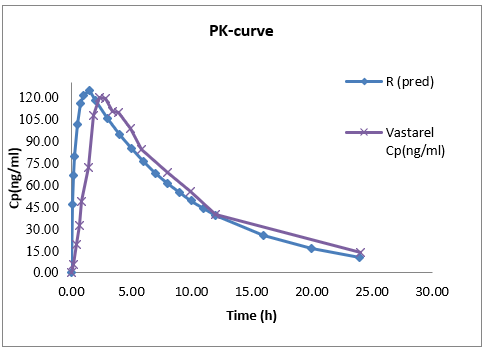
圖6-曲美他嗪PK曲線(文獻值及報道值)
將上述數據進行藥代動力學分析,計算PK參數,結果如下:
|
PK paramters |
預測值 |
文獻值 (Vastarel) |
偏差% |
|
Cmax |
120.05 |
124.62 |
3.812 |
|
AUC 0-t |
1228.78 |
1284.07 |
4.501 |
|
AUC 0-inf_obs |
1366.94 |
1310.50 |
4.128 |
上述數據表明,曲美他嗪體內外模型相關性較好,PK參數預測值與文獻報道值預測誤差低于5%。該模型可成功預測體內吸收。
我方在完成注冊三批樣品后,溶出曲線如圖4所示,根據已建立好模型,對其體內進行預測,所獲得數據較好。因此本品種未進行預BE試驗研究,直接進行BE試驗,空腹條件下,所獲得結果如下表:
|
PK paramters |
T/R(%) |
|
lnCmax |
99.29 94.92-103.86 |
|
lnAUC 0-t |
101.63 98.82-104.51 |
|
lnAUC 0-inf_obs |
101.64 98.96-104.5 |
從溶出曲線可知,鹽酸曲美他嗪片自制品和參比制劑的溶出能夠擬合,體內外相關性模型,較容易建立。但在仿制藥開發過程中,我們通常會遇到自制品很難與參比制劑在各個介質中擬合;不同批次參比的溶出差異較大;由于原料藥溶解性較好,不能找到具有區分力介質等等問題,后續也會針對上述情況,進行系列模型建立的分享。
5. IVIVR未來應用
IVIVR模型是將溶出曲線和體內藥動學行為進行結合,如何確保模型的穩健性,則需要對制劑工藝具有深入的了解及研究,同時也要考慮藥動學參數的影響,如藥物的滲透性、體內的降解等。
IVIVR模型應用逐漸廣泛,不僅可以指導仿制制劑開發,對于新藥開發也具有指導意義,如新藥研發過程中,人體初始劑量的選擇、藥物之間的相互作用等。
參考文獻:
1. USP 23-NF 18; United States Pharmacopeial Convention, Inc., Rockville, MD, 1994.
2. J.E. Polli, J.R. Crison, and G.L. Amidon, Novel approach to the analysis of in vitro-in vivo relationships, J. Pharm. Sci. 85, 753-760 (1996).
3. E. Galia, E. Nicolaides, D. Horter, R. Lobenberg, C. Reppas, and Dressman JB, Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm Res. 15, 698-705 (1998).
4. J.E. Polli, G.S. Rekhi, L.L. Augsburger, and V.P. Shah, Methods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tablets, J. Pharm. Sci. 86, 690-700 (1997).
5.Candy wang, IVIVR在劑型設計中應用,新領先藥訊
6.Sally A. Helmy and Noha O. Mansour. In vitro dissolution and in vivo bioequivalence evaluation of two brands of trimetazidine tables, Clinical Pharmacology in Drug Development. 3(2)139-143.
關于我們:
體內行為與體外行為進行橋接,利用軟件建立體內外相關模型,明確藥學開發目標,預測BE成功率,縮短藥物研發時間,節約產品開發成本,為客戶每個項目順利申報提供保障。
本平臺也針對客戶在BE試驗設計和結果所遇到的問題,提供技術支持,并提供合理、科學的建議。后續著重模型開發,擴大模型應用,為新藥研發提供科學依據。本平臺擁有或計劃擁有全球應用最廣泛的模型模擬軟件和藥動學分析軟件,包括DDDPlus、ADMET predictor、Gastroplus、DAS、PASS、Phoenix WinNonlin等多種軟件。
已經成功對近200個口服固體制劑項目開展了包括藥學目標設定、預BE及正式BE的預測和結果分析解讀等研究工作,其中進入到BE階段的近百個項目,一次性通過率超過80%。此外,平臺還對外承接了幾十個BE預測及結果分析解讀的項目,均精準剖析了客戶于BE試驗過程中存在的問題,并提供了合理完善的指導方案。
體內外橋接平臺負責人為hemanth Joshi先生,曾擔任世界排名前十通用名藥企Dr.Reddy‘s Laboratories、Sun Pharmaceuticals等公司的高級科學家,擁有近20年的藥物制劑研發及臨床評價經驗;曾參與幾十個FDA、EMA注冊ANDA、505b(2)的研發工作,其豐富的工作經歷和卓越的工作能力,帶領新領先醫藥研發水平更上一層樓。
體內外橋接平臺服務范圍
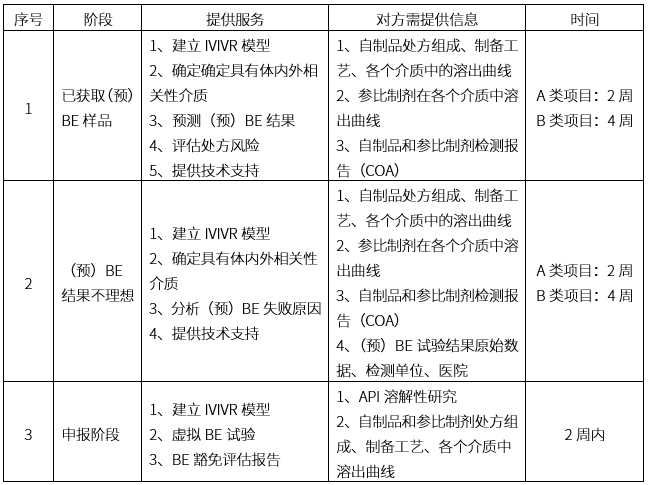
(備注:1.A類項目:普通速釋制劑 ;B類項目:特殊緩控釋制劑 ;2.時間計算為獲得全部數據后,報告提供的時間)
轉載聲明:未經本網或本網權利人授權,不得轉載、摘編或利用其他方式使用上述作品。已經本網或本網權利人授權使用作品的,應在授權范圍內使用,并注明“來源:新領先醫藥科技”。

 010-61006450
010-61006450 聯系地址:
聯系地址: 技術市場部:
技術市場部: 010-61006450
010-61006450
