 Hotline服務(wù)熱線:010-61006450
Hotline服務(wù)熱線:010-61006450
 簡(jiǎn)體中文
簡(jiǎn)體中文劃重點(diǎn):創(chuàng)新藥藥學(xué)開(kāi)發(fā)的技術(shù)要點(diǎn)考量!
編者按:
2015年以來(lái),國(guó)家先后發(fā)布了《關(guān)于改革藥品醫(yī)療器械審評(píng)審批制度的意見(jiàn)》(國(guó)發(fā)[2015]44號(hào))、《關(guān)于深化審評(píng)審批制度改革鼓勵(lì)藥品醫(yī)療器械創(chuàng)新的意見(jiàn)》(廳字[2017]42號(hào))等鼓勵(lì)藥品創(chuàng)新的政策文件,國(guó)內(nèi)創(chuàng)新藥的研發(fā)事業(yè)快速成長(zhǎng),創(chuàng)新藥的申報(bào)數(shù)量明顯增加。
2016-2020 國(guó)內(nèi)創(chuàng)新藥IND、NDA申請(qǐng)數(shù)量
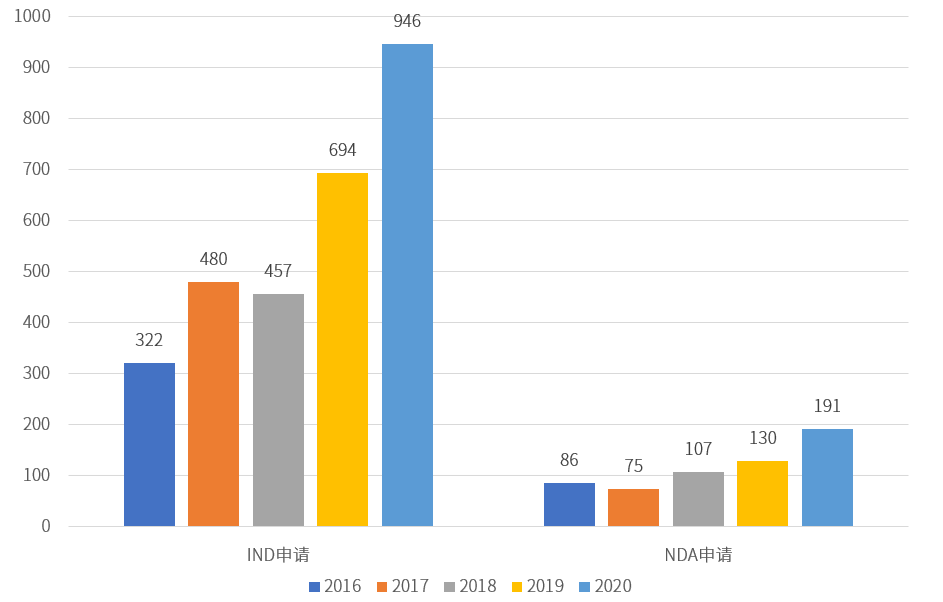
為促進(jìn)創(chuàng)新藥研發(fā)進(jìn)程,解決研發(fā)過(guò)程中存在的可能問(wèn)題,本文針對(duì)創(chuàng)新藥藥學(xué)研發(fā)策略、各階段技術(shù)要點(diǎn)等方面進(jìn)行討論。
創(chuàng)新藥的定義
2020年《藥品注冊(cè)管理辦法》(總局令第27號(hào)):首次明確定義化學(xué)藥品注冊(cè)分類1類為創(chuàng)新藥,指含有新的結(jié)構(gòu)明確的、具有藥理作用的化合物,且具有臨床價(jià)值的藥品。
2020年版《藥品注冊(cè)管理辦法》:
-
將化學(xué)藥品分為創(chuàng)新藥、改良型新藥、仿制藥三大類,并明確規(guī)定了三類藥物的研發(fā)總體要求。
創(chuàng)新藥應(yīng)是具有明確臨床價(jià)值的全球新藥物,改良型新藥應(yīng)是具有明顯臨床優(yōu)勢(shì)的全球新藥物,仿制藥應(yīng)具有與參比制劑一致的質(zhì)量和療效。
-
特別明確的支持以臨床價(jià)值為導(dǎo)向的藥物創(chuàng)新,為鼓勵(lì)真正的創(chuàng)新, 針對(duì)不同的申報(bào)階段給與相應(yīng)的政策和技術(shù)支持,例如技術(shù)指導(dǎo)、溝通交流,優(yōu)先配置資源,縮短審評(píng)時(shí)限,溝通交流后補(bǔ)充提交技術(shù)資料等,制定了4種給予政策優(yōu)惠的藥品審評(píng)審批程序:
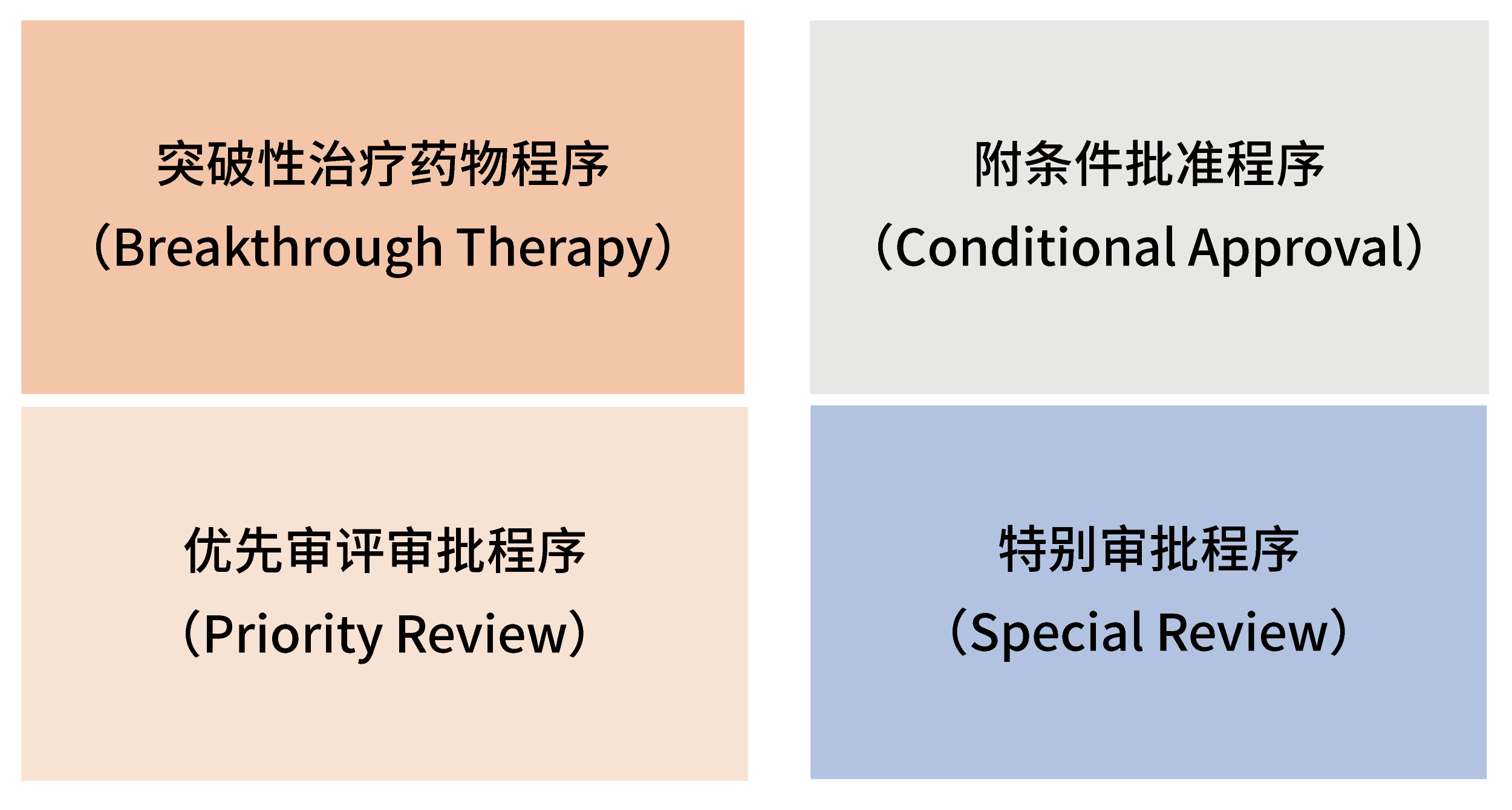
藥學(xué)研究特點(diǎn)
多年來(lái),國(guó)內(nèi)藥品研發(fā)以仿制藥為主,因此不可避免的存在以仿制藥的慣性思維開(kāi)展創(chuàng)新藥研發(fā)的情況:
-
早期藥學(xué)開(kāi)發(fā)工作做得“過(guò)多”、“過(guò)細(xì)”,影響臨床開(kāi)發(fā)的進(jìn)度;
-
研究工作缺乏系統(tǒng)研發(fā)策略和計(jì)劃,不同臨床階段之間的藥學(xué)研究工作是割裂的,不同學(xué)科之間缺乏溝通協(xié)調(diào);
-
對(duì)于風(fēng)險(xiǎn)的識(shí)別和控制能力不足。
〈1〉創(chuàng)新藥與仿制藥具有不同的研發(fā)路徑:
-
創(chuàng)新藥研發(fā)是一個(gè)探索性的研究過(guò)程,是由未知開(kāi)始,基于未被滿足的臨床需求,開(kāi)展藥物篩選與發(fā)現(xiàn)的研究工作;
-
仿制藥則是以參比制劑作為研究標(biāo)桿,目標(biāo)就是要做到與參比制劑的藥學(xué)等同、生物等效。
〈2〉創(chuàng)新藥與仿制藥具有不同的規(guī)律和特點(diǎn):
創(chuàng)新藥的藥學(xué)研發(fā)呈現(xiàn)不同于仿制藥的特有規(guī)律和研究特點(diǎn),具體可歸納為漸進(jìn)性、不確定性。

藥學(xué)研發(fā)策略
基于漸進(jìn)性、不確定性的藥學(xué)研發(fā)特點(diǎn),應(yīng)循序漸進(jìn)的安排藥學(xué)研發(fā)工作。
-
在IND申請(qǐng)階段,主要以臨床試驗(yàn)受試者的安全性為重要考量;
-
臨床試驗(yàn)期間,階段性的藥學(xué)研究工作應(yīng)可支持對(duì)應(yīng)的臨床研究?jī)?nèi)容,并關(guān)注變更前后樣品質(zhì)量的可銜接性;
-
NDA時(shí)再依據(jù)數(shù)據(jù)積累,建立上市藥品質(zhì)量控制體系。
應(yīng)綜合考慮臨床研究?jī)?nèi)容來(lái)制定藥學(xué)研究方案。基于臨床研究的階段(早期臨床研究階段&關(guān)鍵臨床研究階段)、受試人群和研究周期(健康受試者&特定受試者)、品種特點(diǎn)(小分子化合物&復(fù)雜化合物、普通制劑&特殊制劑)、已暴露的和潛在的風(fēng)險(xiǎn),藥學(xué)研究的內(nèi)容以及側(cè)重點(diǎn)也是有所差異的。
此外,應(yīng)根據(jù)藥物開(kāi)發(fā)總體策略制定相應(yīng)藥學(xué)研究計(jì)劃,協(xié)調(diào)開(kāi)展相關(guān)工作,避免藥學(xué)研究與其它研究工作割裂進(jìn)行,同時(shí),藥學(xué)內(nèi)部各專業(yè)(合成、制劑、分析)之間也應(yīng)充分的溝通和配合。
分階段的技術(shù)考慮

〈1〉I期臨床的藥學(xué)研究:
對(duì)于I期臨床申請(qǐng),保證受試者的安全是重要的評(píng)估標(biāo)準(zhǔn),藥學(xué)研究重點(diǎn)關(guān)注與安全性相關(guān)的問(wèn)題,包括雜質(zhì)、穩(wěn)定性、無(wú)菌制劑的生產(chǎn)條件和滅菌/除菌方法等。
由于成藥性尚未確定,該階段的處方工藝一般比較簡(jiǎn)單,無(wú)需考慮工業(yè)化生產(chǎn)的可行性,批量滿足臨床試驗(yàn)以及后續(xù)留樣的需求即可,用于人體試驗(yàn)樣品的生產(chǎn)應(yīng)參照《藥品生產(chǎn)質(zhì)量管理規(guī)范》;該階段尚不用執(zhí)行ICH Q3A和Q3B,雜質(zhì)水平不得超出動(dòng)物安全性試驗(yàn)數(shù)據(jù)所支持的相應(yīng)的雜質(zhì)水平;應(yīng)參照ICH M7,對(duì)樣品中可能存在的致突變雜質(zhì)開(kāi)展研究控制;已有的穩(wěn)定性研究數(shù)據(jù)以及穩(wěn)定性研究方案應(yīng)可支持樣品質(zhì)量在計(jì)劃的臨床試驗(yàn)研究期間符合要求。
〈2〉II/III期臨床的藥學(xué)研究:
對(duì)于II/III 期臨床階段,隨著研究的深入,藥學(xué)研究?jī)?nèi)容要比I期豐富很多,安全性依然是藥學(xué)評(píng)估的重點(diǎn),包括持續(xù)更新的與安全性相關(guān)的問(wèn)題,如雜質(zhì)、穩(wěn)定性等方面的數(shù)據(jù);隨著批量的放大,需要考慮工藝放大的可行性,逐步完善處方工藝;積累與有效性相關(guān)的信息,如晶型、粒度、釋放行為等;基于歷史批次(尤其是安全性試驗(yàn)批次、關(guān)鍵臨床試驗(yàn)批次)生產(chǎn)信息、質(zhì)量特性、穩(wěn)定性研究結(jié)果等,完善質(zhì)量標(biāo)準(zhǔn)、包裝、貯藏條件等。
〈3〉NDA的藥學(xué)研究:
對(duì)于 NDA,藥學(xué)研究需要遵循國(guó)內(nèi)及ICH等已發(fā)布的相關(guān)技術(shù)指導(dǎo)原則。藥學(xué)研究的重點(diǎn)是基于歷史批次的生產(chǎn)數(shù)據(jù)和批分析數(shù)據(jù),對(duì)上市藥品質(zhì)量控制體系進(jìn)行全面評(píng)估,確保擬上市的藥品具有與關(guān)鍵臨床試驗(yàn)樣品一致的/持續(xù)穩(wěn)定的質(zhì)量。
仿制藥可參考原研產(chǎn)品的相關(guān)信息建立質(zhì)控要求,而創(chuàng)新藥則需要根據(jù)研發(fā)過(guò)程中積累的試驗(yàn)數(shù)據(jù)來(lái)建立藥品有效安全性與產(chǎn)品質(zhì)量的關(guān)聯(lián),因此需要注意試驗(yàn)數(shù)據(jù)的完整、詳實(shí),特別是關(guān)鍵Ⅲ期臨床批次的數(shù)據(jù)。
※ 應(yīng)特別關(guān)注與藥品安全有效性相關(guān)的關(guān)鍵質(zhì)量屬性,例如有關(guān)物質(zhì)、粒度、晶型、溶出行為等,要注意臨床前安全性批次、臨床試驗(yàn)批次、商業(yè)批、穩(wěn)定性批次產(chǎn)品質(zhì)量的相關(guān)性。
創(chuàng)新藥研發(fā)過(guò)程中的變更
在創(chuàng)新藥的研發(fā)過(guò)程當(dāng)中,變更是一個(gè)永恒的主題,是不可避免的。但這并不意味著,在每個(gè)研究階段都頻繁的發(fā)生變更,變更一般主要是發(fā)生在I、II期臨床研究階段。
因?yàn)镮II期是全面的驗(yàn)證藥品安全性、有效性的關(guān)鍵臨床試驗(yàn),越是后期的變更對(duì)產(chǎn)品開(kāi)發(fā)的影響就越大,需要開(kāi)展的研究驗(yàn)證工作就越多,因此對(duì)于創(chuàng)新藥藥學(xué)開(kāi)發(fā)計(jì)劃的制定,無(wú)論是從時(shí)間成本、還是經(jīng)濟(jì)成本的角度來(lái)考慮,一般不會(huì)把重大的變更放到后期來(lái)做。
應(yīng)盡量避免III期臨床期間發(fā)生影響產(chǎn)品質(zhì)量的重大變更,在這個(gè)階段即使需要變更,一般也都是一些小的調(diào)整,例如,工藝的微調(diào)、優(yōu)化質(zhì)量標(biāo)準(zhǔn)、嚴(yán)格貯存條件等。
應(yīng)結(jié)合品種特點(diǎn)和變更內(nèi)容、遵循風(fēng)險(xiǎn)評(píng)估的思路,評(píng)估變更對(duì)樣品質(zhì)量的影響,以及進(jìn)一步的對(duì)臨床試驗(yàn)受試者安全性、臨床試驗(yàn)結(jié)果科學(xué)性的可能影響。同樣,評(píng)估時(shí)應(yīng)充分考慮變更的階段、受試人群、品種特點(diǎn)、認(rèn)識(shí)的局限性等。根據(jù)影響的大小,可將變更分為重大變更和一般變更,重大變更應(yīng)提出補(bǔ)充申請(qǐng),一般變更可體現(xiàn)在安全性更新報(bào)告中。
結(jié)語(yǔ):創(chuàng)新藥研發(fā)有其自身規(guī)律和研發(fā)思路,研究工作應(yīng)按照其規(guī)律有序開(kāi)展。在創(chuàng)新藥的開(kāi)發(fā)過(guò)程中,安全性和有效性問(wèn)題是淘汰候選藥物的主要原因,藥學(xué)研究的目的是要支持其安全性和有效性的評(píng)價(jià),為臨床研究、動(dòng)物研究提供恰當(dāng)?shù)难芯坑脴悠贰?yīng)根據(jù)產(chǎn)品的整體開(kāi)發(fā)策略、品種特點(diǎn),基于藥學(xué)研究的漸進(jìn)性、不確定性的特點(diǎn),科學(xué)合理的針對(duì)性制定藥學(xué)研究方案。
-END-
關(guān)于我們:
北京新領(lǐng)先(股票代碼:600222)成立于2005年,是一家面向全球提供藥學(xué)臨床前研究、臨床CRO和CDMO服務(wù)的高新技術(shù)企業(yè),連續(xù)多年被評(píng)為“中國(guó)醫(yī)藥研發(fā)公司10強(qiáng)(2019年位列第一)”。公司總部位于北京中關(guān)村高新技術(shù)園區(qū),同時(shí)在鄭州臨空生物園區(qū)建立了新藥篩選及檢測(cè)平臺(tái)、藥物評(píng)價(jià)平臺(tái)(動(dòng)物房,GLP、AAALAC、CNAS認(rèn)證)、大分子中試及大規(guī)模生產(chǎn)服務(wù)平臺(tái)、小分子CMC制劑研究生產(chǎn)平臺(tái)、細(xì)胞技術(shù)服務(wù)平臺(tái)和臨床CRO平臺(tái)等六大符合國(guó)際標(biāo)準(zhǔn)(FDA、EMA和NMPA GMP標(biāo)準(zhǔn))的研發(fā)平臺(tái),形成“新領(lǐng)先CXO”全產(chǎn)業(yè)鏈服務(wù)體系。仿創(chuàng)結(jié)合,雙引擎驅(qū)動(dòng),能夠?yàn)榭蛻籼峁┧帉W(xué)研發(fā)全生命周期的多元化服務(wù)。


轉(zhuǎn)載聲明:未經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán),不得轉(zhuǎn)載、摘編或利用其他方式使用上述作品。已經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán)使用作品的,應(yīng)在授權(quán)范圍內(nèi)使用,并注明“來(lái)源:新領(lǐng)先醫(yī)藥科技”。

 010-61006450
010-61006450 聯(lián)系地址:
聯(lián)系地址: 技術(shù)市場(chǎng)部:
技術(shù)市場(chǎng)部: 010-61006450
010-61006450
