 Hotline服務熱線:010-61006450
Hotline服務熱線:010-61006450
 簡體中文
簡體中文收藏!緩釋微球制劑的質量控制
所謂藥物緩釋,是指使藥物在生物體內緩慢釋放或控制藥物以一定速度釋放,以達到其在特定部位釋放或按預設速度釋放的目的。例如現在研究較為廣泛的載藥微囊緩釋系統以及載藥微球緩釋系統等,均具有藥物緩釋體系的典型優良特性。
微球(microsphere)是指藥物分散于或被吸附在聚合物基質、高分子中而形成的一種微粒分散體系。微球作為一種應用較為普遍的新型給藥系統,其粒徑一般分布在 1~250µm 的范圍內,粒徑在10~1000nm 之間的稱納米球。該技術開發的制劑可用于肌肉注射、動靜脈注射、粘膜給藥、關節腔內給藥、口服以及皮內注射等幾乎各種方式的給藥途徑。
國外對載藥微球的研究開始較早并正在逐步深入,20 世紀八九十年代,一些研究機構如美國佛羅里達大學、法國巴黎第十一大學、日本秋田大學和日本城西大學等都設立有專門的載藥微球研究室,近些年來,也有成型的載藥微球商品(如淀粉微球)出售。我國對載藥微球的研究開始時間相對較晚,雖然已取得了一些初步成果,但技術還不成熟,上市藥品較少。
相比于傳統的劑型,微球制劑的主要優點在于:
?掩蓋藥物的不良氣味及口味;
?提高藥物的穩定性,防止藥物在體內失活;
?通過載體材料及制備技術的選擇,可實現藥物在體內的緩慢持續釋放,減輕患者頻繁多次給藥的痛苦,提高患者順應性;
?可使藥物濃集于靶區,提高療效,減少藥物的毒副作用;
?除藥物外,還可將活細胞包埋于載體內。
緩釋微球制劑在臨床上的釋藥周期往往長達數周,甚至數月,一旦不能保證產品的質量(如突釋過高,釋放周期難以控制,載體材料的安全性等方面),會導致發生毒副作用甚至威脅患者的生命,所以對于微球制劑應該具有比常規制劑更加嚴格的質量要求和批次間的重復性要求。
我們結合各指導原則及研發經驗,確定以下方面需要關注:目前,各國藥典采取指導原則的形式對微球制劑的質量評價提出了要求和規定,如微球指導原則首次出現在15版藥典中,并在2020版進行提升,提出了“微粒制劑指導原則”,對國內進行緩釋微球開發的藥企提出了質量控制的導向。
通過掃描電子顯微鏡(SEM)或者透射電子顯微鏡(TEM)觀察微球的形態,如形狀(圓形或類圓形)、表面形貌(光滑或粗糙)、骨架結構(多孔或實心)(圖1)。理想微球的微觀形態應為圓整球型或橢圓形實體,形態飽滿,顆粒的大小應盡可能均勻,微球之間無粘連。微球形態與結構的不同對微球的載藥量以及釋放行為有顯著影響。表面粗糙的微球易吸附藥物結晶,往往會導致高突釋。通過對微球形態進行觀測,總結形態與處方工藝之間的關系,不但可以對微球的制備機理進行探索,還可以對釋放行為進行優化。

圖1 電鏡下不同形態的微球
SEM是目前觀察微球形態使用最廣泛的方法,被用于表面及切面形態的觀察(見圖2)。TEM 分辨率高,圖像為二級結構平面,適用于亞微球、納米球粒徑測定。此外還有原子力學顯微鏡(atomic force microscopy,AFM)可用于觀察微球形態,AFM 優點之一是分辨率高,與 SEM 相比,不需要對樣品進行金屬噴鍍,避免了噴鍍后對樣品的表面形態造成的破壞,并且AFM 允許在液態環境下觀測樣品,而 SEM 則不行。但是 AFM 缺點是觀察范圍窄,得到數據不具有統計性,適合單個粒子表面形態的觀察。

圖2 SEM 觀察的 PLGA-Glu 微球圖像
(A)全視圖 (B)單個微球表面圖 (C)單個微球切面圖
粒徑及粒徑分布是影響微球制劑釋放行為的關鍵因素,其對緩釋微球的包封率、釋放行為模式、降解速率都有一定影響,因此粒徑大小及分布是質量控制中一個很重要的指標。隨著檢測手段的進步,粒徑大小及其分布測定由傳統的光學顯微鏡視野測定發展到電子顯微鏡的逐一統計再發展到近年來應用廣泛的激光粒度儀繪制粒徑大小分布圖,可以在制備過程中的每一步工藝里對粒徑大小及分布進行追蹤檢測進而得到目標微球,表1介紹了各種方法的特點。
表1 常用微球的粒度及粒度分布檢測方法
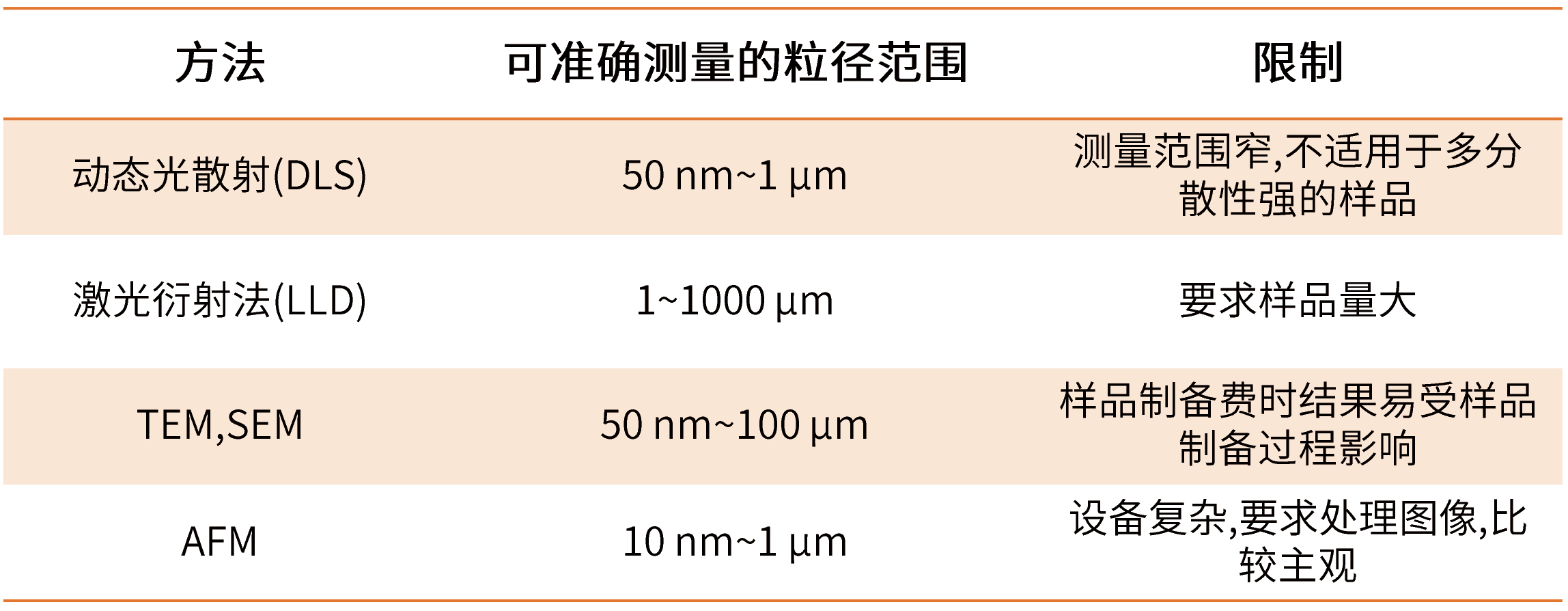
而 粒 徑 的 分 布 除 了 可 用 粒 徑 分 布 圖 表 示 , 還 可 用 多 分 散 性 指 數(polydispersity index,PDI)和跨距表示。跨距與多分散性指數數值越小,表示粒徑分布越均勻。
由于微球制劑與普通制劑不一樣,除了必要的輔料,在生產過程中還可能會使用二氯甲烷、正庚烷、乙醇或乙酸乙酯等有機溶劑,制備過程中引入的油相(有機溶劑)在固化的過程中會存在未能完全除去的問題,諸如丙酮、乙酸乙酯、二氯甲烷等的殘留不僅影響微球儲存的穩定性,還會在注射后引起人體的副作用,因此每個國家的藥典都對微球的有機溶劑殘留量有著嚴格的要求。依據 ICH 指導原則分類,二氯甲烷屬于第二類殘留溶劑,而正庚烷、乙醇和乙酸乙酯為第三類殘留溶劑,因此必須對其限度進行控制。
此外,對于固體無菌粉末制劑,一般都要求控制水分的含量。由于微球制劑大都是多肽或蛋白類藥物,這類藥物對熱不穩定,因此不適合采用干燥失重的方法測定水分,可以采用卡爾-費休氏水分測定方法來完成。
載藥量和包封率是反映微球制劑中藥物含量的重要指標,載藥量的批間穩定性也是工藝成熟的重要標志。
載藥量是指微球制劑中所含藥物的質量分數,其檢測方法一般是先采用合適的有機溶劑將微球高分子材料骨架溶解,再根據藥物的性質選擇不同的方法將藥物分離或提取出來,進行含量測定。
包封率是指微球制劑中包封的藥量占微球制劑中包封與未包封總藥量的比值,其測定先要將微球粉末溶于注射用溶劑,再通過離心法、過濾法、凝膠柱色譜法分離后測定。
二者是衡量制備工藝和成本的重要指標。載藥量和包封率的計算都需要建立在藥物含量測定的基礎上,目前上市的微球制劑所用載體多為聚乳酸-羥基乙酸共聚物(PLGA)。由于 PLGA 易溶于二氯甲烷、三氯甲烷、二甲亞砜等有機溶劑,而不溶于水、醇。依據藥物和 PLGA 的溶解性質,PLGA 微球常用的含量測定方法有:
① 先用有機溶劑溶解 PLGA 和藥物,再用不溶于 PLGA 溶劑沉淀 PLGA,經過離心或過濾后,取上清進液相測定含量;
② 先用有機溶劑溶解 PLGA 和藥物,再加入醋酸鹽緩沖液等溶劑提取多肽后進樣分析;
③ 溶劑溶解 PLGA 及藥物后,直接進樣測定。
方法比較:
方法①和②需要在測定之前將高聚物與藥物分離,而且分離過程使用的試劑容易導致藥物損失。方法③的優勢在于無需將高聚物與藥物分離,但是應用較少,需要使用質譜等特殊儀器。
釋放行為是根據臨床適應癥需求和高分子聚合物材料性質共同決定的。選擇合適的高分子聚合物材料與工藝制備不同結構的載藥微球,使活性成分按照預期的藥代動力學模型釋放。
-
釋放速率
對于可生物降解材料,溶脹和溶蝕機制也是控制藥物釋放的主要因素。釋放介質的組成、pH 值、離子強度、滲透壓和溫度等都會對釋放速率產生影響。
在載藥微球的研發階段,應確定好合適的體外釋放條件,并根據體內釋放條件建立體內、體外相關性。對于釋放周期較長的載藥微球,可以建立加快釋放試驗的方法,預測模擬常規釋放行為。建立加速釋放的條件要遵循相關性原則,使加速釋放曲線盡量擬合常規釋放曲線,得到準確的相關性。
-
突釋效應
同時需要注意的是載藥微球的突釋效應,在微球釋放的最初階段,吸附在微球表面的藥物會通過擴散作用而快速釋放,稱為突釋效應。由于微球表面吸附的藥物大量釋放,短時間內使局部藥物濃度快速升高,極易引起副作用。
突釋效應可能導致人體內藥物濃度在短時間內迅速升高,并使得藥物效期縮短,是限制微球廣泛應用的關鍵問題,因此在質量控制過程中必須重點關注突釋率這一指標。
2020 版《中國藥典》明確規定載藥微球在前 0.5h 內釋放的藥物含量要低于40%,收載的釋放度測定法包括槳法、籃法、杯法,用以上方法測定釋放度不僅需要大量的樣品,而且釋放后測得的藥物濃度偏低,已無法滿足檢驗要求。而對于緩釋微球制劑的體外釋放方法,目前并沒有統一的標準要求,目前報道的微球制劑體外釋放度測定方法主要有:
① 直接釋藥法,這是目前最常用的方法,包括搖床法和恒溫水浴靜態法。微球制劑置含有介質的容器中保持恒溫封閉,一定時間取樣并補充新鮮介質。
② 流通池法,系統由恒流泵、溫控流通池、存儲瓶、過濾系統、取樣系統和樣品收集系統組成。此法已被美國藥典收載,被廣泛應用于緩釋制劑的研究。
③ 透析膜擴散法,該法是指將微球放入透析管中,并將其放入介質中測定。3種方法優缺點比較見表2。
表2 微球制劑體外釋放度實驗方法的比較

除了改進體外釋放度的實驗裝置,還可以通過調節釋放介質溫度、pH 值、離子強度、攪拌速率以及使用表面活性劑、酶等方式能實現微球體外加速釋放,而達到縮短檢驗周期,提高檢驗效率的目的。
鑒于已上市的產品大部分為多肽微球,其相關雜質包括降解雜質、工藝雜質以及聚合物雜質:
? 降解雜質包括藥物在生產、儲存過程中發生水解、氧化反應而生成的產物。
? 工藝雜質中得到最廣泛關注的是乙酰化雜質,這類雜質是由藥物多肽中的氨基、羥基與 PLGA 的羧基末端經過化學反應生成的,這種雜質目前在 PLGA 微球中廣泛存在。乙酰化雜質的產生與多肽自身結構有關,質譜和毛細管電泳技術聯合使用可用于其他多肽微球的乙酰化雜質檢測。
? 聚合物雜質包括多肽或蛋白自身相聚合形成的雜質,以及聚合物-多肽雜質。
緩釋微球的微生物檢查比一般凍干制劑要求更加嚴格,這是因為在微球的制備和生產過程中,眾多環節很有可能引入微生物,而微球的尺寸以及高分子材料的特性使微生物更易吸附在其表面,也可以被包裹在骨架內部,所以要在微球的內部以及外部根據不同的制備條件對內毒素和無菌作系統全面的篩查。微球制劑的細菌內毒素和無菌檢查,需要進行球內和球外部檢測實驗。
目前,針對已上市的微球,可以利用一定濃度的二甲基亞砜將微球溶解和破碎,將溶解后全部液體接種至培養基中,運用顯微鏡觀察溶解和培養過程。
Zeta電位也是微球的一個重要屬性,Zeta電位往往能指征微球制劑的穩定性,而這一指標卻容易被忽視。在微粒分散體系的溶液中,其表面帶有同種離子,通過靜電引力吸附和擴散作用,在微粒周圍形成的吸附層與相鄰的擴散層共同構成微粒的雙電層結構,從吸附層表面至反離子電荷為零處的電位差叫動電位,即Zeta電位。Zeta電位值可以反映微粒的物理穩定性,Zeta電位越大,微粒之間的排斥作用越強,絮凝或沉積的可能性越小,微粒在溶液中越穩定。一般ζ電位絕對值大于15mV,可以達到穩定性要求。
目前市場上測量Zeta電位已有專門的Zeta電位儀,英國馬爾文和美國貝克曼公司都已推出電位儀系列產品,直接進樣就可以讀出Zeta電位值。
微球制劑的緩釋功能是通過載體輔料實現的,這些載體輔料通常無毒、可降解并具有良好生物相容性。常用的微球制劑載體輔料包括天然材料(如明膠、殼聚糖、淀粉、白蛋白),半合成材料(多為纖維素衍生物)以及合成材料(聚乳酸、聚氨基酸、聚羥基丁酸酯、聚乳酸-羥基乙酸共聚物等)。此外,在微球生產過程還需要加入乳化劑、潤濕劑以及表面活性劑等輔料。載體輔料的分子量及分布范圍、組成單體的比例、以及玻璃轉化溫度(glass transition temperature,Tg)都會影響微球的釋放周期、釋放速度。
分子量及其分布測定主要采用凝膠滲透色譜法(GPC),并以重均分子量(Mw)、數均分子量(Mn)和分子量分散系數(Mw/Mn),或者繪制分子量分布曲線來表征其分子量。
在微球藥物的釋放過程中,藥物的釋放伴隨著聚合物骨架的水解。聚合物降解的速率決定藥物釋放的速率。因此可以在釋放過程中觀察微球的形態,并通過凝膠滲透色譜法(GPC)檢測不同時刻高分子聚合物載體的分子量,通過形態及分子量的變化監控微球降解釋放過程。這些信息對于篩選骨架材料、優化制備工藝有著重要意義。
玻璃轉化溫度(Tg)是載體輔料在玻璃態和高彈態之間相互轉化的溫度,在一定程度上可以反映微球的穩定性。Tg 可以通過差示掃描量熱分析法進行測定。在質量控制過程中,關注載體輔料分子量等特性變化,可以幫助我們理解微球降解機理,優化處方工藝。
在高分子聚合物材料析出形成微球后,聚合物的Tg會發生改變。因為當聚合物和藥物或溶劑共同存在時,易產生共價鍵吸引力,使得聚合物的Tg降低。比較常用的 Tg 檢測方法是差示熱分析法(DTA)和差示掃描量熱法(DSC)。高分子聚合物的晶型與結晶度的變化可以從側面反映藥物的釋放速率和微球的降解速度。可利用X射線衍射法檢測聚合物結晶度從而佐證載藥微球的釋放行為與規律。
制備載藥微球過程中,微球的載藥量和包封率受許多因素的影響,主要的因素有:PLGA 的組成、主藥的理化性質、各溶劑相的用量、微球制備方法及制備參數等。
-
PLGA 的組成:PLGA 的相對分子質量和乳酸/羥基乙酸(LA/GA)比例可控,選用不同的PLGA 制備微球可得到不同的載藥量和包封率。
-
PLGA 在第一相中的濃度
微球的載藥量隨著 PLGA 濃度增加而提高。可以從 3 個方面解釋這個現象:
① 高濃度的聚合物可以加速微球的固化,并可有效阻止藥物向外相的擴散;
② 高濃度的聚合物可以增加有機相的黏度,從而減緩小液滴中藥物的擴散;
③ 高濃度的聚合物制備出相對較大粒徑的微球,在清洗微球表面藥物時較小粒徑微球損失的藥物要少。
-
PLGA 在有機溶劑中的溶解性
PLGA 在有機溶劑中的溶解度決定了微球的固化速率,較高的溶解度會延長微球固化時間,導致更多的藥物擴散到連續相中,降低包封率。一般來說,相對高的 LA/GA 比例組成、低分子質量、末端閉合的 PLGA 親水性較差,在二氯甲烷中的溶解性較好,選用此類型的 PLGA 制備的微球包封率較低。此外,PLGA 的親水性還可增加初乳的穩定性,從而獲得高包封率。
-
藥物理化性質的影響
藥物的理化性質決定溶劑系統的組成及制備方法的選擇。通常疏水物質宜選用 O/W 法,親水性物質選用 O/O 和 W/O/O 法可以獲得高包封率;水溶性化合物,如在有機溶劑中不穩定的物質(如蛋白質和多肽類),宜選用 W/O/W 技術。若藥物為多肽或氨基酸、酶等,還應考慮其在微球制備過程中的穩定性。為了增加蛋白質的穩定性,可以考慮在微球制備過程中加入兩親性穩定劑、堿性鹽或者凍干保護劑等。
-
藥物與聚合物的比例
提高聚合物和藥物的比例可有效阻止微球制備過程中藥物向外相的滲漏,從而獲得高的包封率。
-
藥物與聚合物之間的作用
藥物與聚合物之間的作用也會影響微球的包封率。通常,親水性藥物與末端含有親水基團的 PLGA 結合所得微球包封率較高,而疏水性藥物與相對疏水的末端閉合的 PLGA 結合所得微球的包封率較高。
-
藥物在第二相中的溶解性
微球的固化過程是造成藥物損失的一個重要環節。如果藥物在第二相中的溶解度大于在第一相中的溶解度,將更容易擴散到第二相中,導致微球包封率降低。用 W/O/W 法制備親水性蛋白質類藥物微球時,可通過兩種方法提高親水性藥物微球包封率:
① 調節第二相的 pH值、滲透壓(加鹽)等,使蛋白質溶解性降低。pH 值影響蛋白質的溶解度,隨著 pH值的降低,蛋白質的溶解度也降低;
② 制備蛋白質的包合物或 Zn 復合物,降低其在第二相中的溶解度。
-
溶劑的影響
溶劑的理化性質,如沸點、揮發性及與其他溶劑的互溶性等因素,關系到藥物或者聚合物在溶劑中的溶解度以及溶劑去除的速率。
? 溶劑種類和組成
有機溶劑對微球的包封率和釋放有顯著影響。采用復乳法制備蛋白微球時,通常選擇二氯甲烷、乙腈、乙酸乙酯、丙酮、二甲亞砜等有機溶劑作為溶解聚合物的介質。為了改變藥物或者聚合物在溶劑中的溶解度,可以在溶劑中加入其他溶劑改變其理化性質。在有機相中加入能與水混溶的有機溶劑如丙酮、甲醇、乙酸乙酯、二甲基亞砜等,可以提高水溶性藥物的溶解度,提高微球的載藥量和包封率。在內相中加入極性溶劑可以加快萃取過程,促進微球的快速形成,抑制藥物向外水相的擴散,從而提高微球的包封率。
? 溶劑體積
使用乳化溶劑揮發法制備微球時,第一相和第二相的比例是微球載藥量的重要影響因素。降低第一相/第二相比例能顯著增加微球載藥量和包封率,減少突釋。因為增大第二相的量可達到稀釋溶劑的效果,從而使有機溶劑以高濃度梯度跨過相界面。
? 溶劑去除速率
當藥物易分配到連續相中時,溶劑去除速率就成為一個關鍵影響因素。在乳化-溶劑揮發或者乳化溶劑萃取法中,有機溶劑可以在其沸點附近揮發或者被從第二相萃取出來。前者的速率可以由蒸發溫度控制,后者的速率可以由稀釋介質的量來控制。有研究考察了不同溫度下制得微球包封率情況,發現升高溫度在加快有機溶劑去除速率的同時,也會增加某些藥物在外水相的溶解度,加快向外水相的質量傳遞,而且不適用于高溫不穩定的藥物。
? 添加劑
PLGA 因其固有的疏水性對親水性強的分子的親和力低,所以包封這些分子時的效率不高,造成制備過程中活性物質的大量損失。為提高 PLGA 微球包封率,引入添加劑可以有效解決這一問題。
常用的附加劑有多元醇類(如甘露醇、海藻糖、山梨醇等),非離子表面活性劑(如聚山梨酯20、司盤80、普朗尼克F68等),大分子化合物[人血清蛋白(HSA)、聚乙二醇(PEG)、羥丙基-β-環糊精(HP- β-CD)]等。在復乳溶劑揮發/萃取法中,可以在內外水相或者油相中加入一些物質,如鼠血清白蛋白、吐溫 80、司盤 80、PVA 等來提高初乳穩定性,防止內外水相合并,從而提高包封率。此外,還可以通過調節外水相乳化劑或電解質的濃度的方法,改變藥物和有機溶劑在水相中的溶解度。
一般來說,在外水相加入一定濃度的電解質(如氯化鈉),可以獲得高包封率,可能與內外水相間滲透壓梯度形成有關。
與其他傳統劑型相比,注射用緩釋微球制劑在藥物制劑和臨床診斷/治療領域商品化速度相對緩慢,并且微球制劑從實驗室階段的初期研發走向臨床應用需要經歷漫長的過程,主要有以下幾個瓶頸需要解決:
-
各國藥品監督評審機構并沒有針對緩釋微球制劑推出專門的監管和評審要求;
-
微球制劑的放大生產受到設備自動化的制約,難以大規模、大批量制備,導致成本過高,并且重復性較差;
-
FDA 批準用于臨床的生物可降解高分子聚合物的種類及規格有限;
-
載藥微球進入人體后,降解釋放以及與組織間發生作用的機制機理尚不清晰明確,毒性和安全性評價還需進一步驗證。
近年來,新型給藥系統研究與開發的投入和支持逐年增加,對于蛋白及多肽類藥物的新型緩釋/控釋注射給藥系統的研究也成為熱門。而從實驗室技術取得成功到逐級放大生產發展到產業化規模,其間須逾越層層障礙。如何在保持基礎研究成果的條件下,降低成本,逐步擴大產量,直至產業化生產,也是接下來發展新型給藥系統中緊迫而又重要的任務。
另外,緩釋微球技術長期以來被發達國家的藥企壟斷,直到近十幾年,國內各大藥企才投入大量的人力與資金開展對微球制劑的研究。目前已有亮丙瑞林微球成功上市,曲普瑞林微球和利培酮微球正在臨床試驗中,相信在不久的將來,會有越來越多的微球制劑產品成功走向市場。
-END-
關于我們:
北京新領先成立于 2005 年,注冊資金近億元,于 2015 年實現上交所上市(股票代碼:600222),是一家面向全球提供藥學臨床前研究、臨床 CRO 和CDMO服務的高新技術企業,連續多年蟬聯“中國醫藥研發公司榜首”。
目前,公司已與國內外上下游500余家企業建立合作關系,其中部分企業為戰略合作關系。累計承接了藥學研發項目400余項、臨床研究項目500余項,申請專利200余項,獲得授權近百項。
公司從建立伊始就以“是我,讓中國新藥技術和生產工藝與世界同步”作為企業愿景,致力于用創新的研發和服務模式以及信息化技術推動中國醫藥產業發展,縮小與世界先進水平間的差距,讓中國人民早日用上安全藥和良心藥!
公司總部位于北京中關村高新技術園區,擁有 10000 平米研發實驗室,同時在鄭州臨空生物園區建立了新藥篩選及檢測平臺、藥物評價平臺(動物房,GLP、AAALAC、CNAS 認證)、大分子中試及大規模生產服務平臺、小分子 CMC 制劑研究生產平臺、細胞技術服務平臺和臨床 CRO 平臺等六大符合國際標準(FDA、EMA 和 NMPA GMP 標準)的研發平臺,形成“新領先 CXO”全產業鏈服務體系。仿創結合,雙引擎驅動,能夠為客戶提供藥學研發全生命周期的多元化服務。


轉載聲明:未經本網或本網權利人授權,不得轉載、摘編或利用其他方式使用上述作品。已經本網或本網權利人授權使用作品的,應在授權范圍內使用,并注明“來源:新領先醫藥科技”。

 010-61006450
010-61006450 聯系地址:
聯系地址: 技術市場部:
技術市場部: 010-61006450
010-61006450
