 Hotline服務熱線:010-61006450
Hotline服務熱線:010-61006450
 簡體中文
簡體中文收藏 | 全過程!滴眼液研究內容要點及注意事項
滴眼劑系指由原料藥物與適宜輔料制成的供滴入眼內的無菌液體制劑,其生產和質量要求一般參考注射劑的技術要求。本文結合《化學藥品注射劑仿制藥質量和療效一致性評價技術要求》等相關要求,具體闡述滴眼液研究過程中的研發(fā)內容及注意事項。
- 載入《仿制藥參比制劑目錄》的品種,按照參比制劑目錄進行選擇。
- 尚未收載入《仿制藥參比制劑目錄》的品種,藥品上市許可持有人應當按照《國家藥監(jiān)局關于發(fā)布化學仿制藥參比制劑遴選與確定程序的公告》(2019年第25號)申報參比制劑,待參比制劑確定后開展研發(fā)申報,避免出現(xiàn)因參比制劑選擇與國家公布的參比制劑不符,影響研究項目開展、造成資源浪費等問題。
當擬研發(fā)藥品規(guī)格(包裝規(guī)格)與所公布參比制劑不一致情況,應提出進一步評估風險后再進行開發(fā),如有必要,可同步進行備案。
一般應至少采用3批仿制制劑與參比制劑進行物理化學特性(Q3)對比,用于對比研究的仿制制劑應與商業(yè)化批次在同一生產線上生產。在工藝驗證之前,應有2個批次參比制劑研究結果(最少需要1批研究結果),以便再次確認研發(fā)目標的可靠性。
研究內容包括:
關鍵質量屬性剖析:包括性狀、澄清度與顏色、pH值、裝量或最低裝量、滲透壓摩爾濃度(比)、有關物質(異構體)、含量、抑菌劑含量、穩(wěn)定劑含量(如有)等,同時還需重點對參比制劑黏度、比重(相對密度)、滴出量、緩沖容量進行剖析。
穩(wěn)定性研究:影響因素、低溫凍融、加速、中間、長期等,應注意該類品種包裝為半滲透性包材,需要低濕度穩(wěn)定性箱。考察指標除以上關鍵質量屬性外,還需重點關注失水率。
其他:包材反向剖析,確定包材材質和尺寸;原輔料用量剖析或確認。
實驗室研究前信息確認
通過文獻、上市說明書以及參比制劑剖析結果,對制劑的適應癥、劑型、規(guī)格、用法用量、藥代動力學、有效期、包裝系統(tǒng)等信息進行前瞻性總結,并進行合理性說明,需要調研是否在FDA或EMA有各藥指南(如有)。
藥品質量屬性包括但不限于以下內容:性狀、鑒別、檢查[澄清度、顏色、pH值(眼睛可耐受的pH范圍為5~9)、滲透壓摩爾濃度(與淚液等滲)、裝量、抑菌劑(通常多劑量包裝需加入抑菌劑,單劑量包裝不得加入抑菌劑)、穩(wěn)定劑(如有)、可見異物、不溶性微粒、無菌、有關物質(異構體)、含量等。
? 注意失水率、比重(相對密度)、黏度、滴出量、緩沖容量、抑菌效力的測定,以有效控制滴眼劑的質量。
判定標準:是否屬于關鍵質量屬性取決于當該質量屬性超出可接受范圍時由風險評估獲得的該屬性對臨床有效性和安全性的影響程度和不確定性。內容應當包括:藥品質量屬性,范圍,是否為CQA,合理性說明,質量控制風險級別。
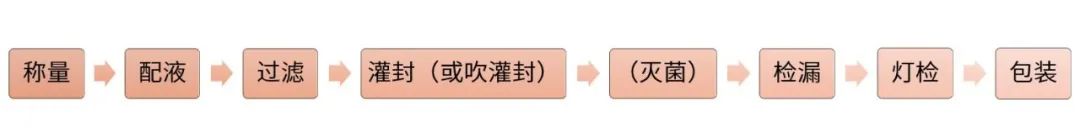
制備工藝流程
工藝參數(shù):
? 配液:是否充氮、配液溫度、加料順序、是否避光、pH調節(jié)范圍、中間體藥液穩(wěn)定性。
? 過濾:過濾材質。
? 滅菌:滅菌參數(shù)(根據(jù)物料性質及包材材質確定)。
? 檢漏:容器密封完整性驗證,確定檢漏參數(shù)。
? 滴眼劑包裝容器應無菌、不易破裂,其透明度應不影響可見異物檢查。除另有規(guī)定外,眼用制劑應遮光密封貯存。眼用制劑(含有抑菌劑)在啟用后最多可使用4周。除另有規(guī)定外,每個容器的裝量應不超過10ml(通則0105)。
實驗室研究
進行原料藥與參比制劑雜質譜對比研究,初步判斷原料藥的質量情況(工藝雜質及降解雜質剖析)并進行原料藥來源篩查。
? 不同溫度、pH下的溶解度和溶解速度(根據(jù)原料藥溶解性確定)。
? 吸濕性曲線考察。
? 穩(wěn)定性研究:若原料藥貯存條件為低溫,以有關物質及水分為指標考察恢復至室溫貯存時限等。
基于參比制劑說明書、原研專利、公開審評報告等披露的處方信息,結合參比制劑輔料用量剖析結果擬定自制品處方。
原輔料來源與廠家提前溝通(原則上選用已有登記號且同意授權的原輔包,如未登記,需確認物料供應商登記和授權情況)。
如有緩沖劑,需對自制品的緩沖能力進行測試,不低于參比制劑。
多劑量眼用制劑一般應加適當抑菌劑,盡量選用安全風險小的抑菌劑(與參比制劑一致),產品標簽應標明抑菌劑種類和標示量。抑菌劑用量參照參比制劑用量加入,并在此基礎上設計低抑菌劑濃度梯度處方(可參照質量標準擬定的抑菌劑含量下限),對抑菌效力進行考察,確保穩(wěn)定性期間的抑菌劑含量具有足夠的抑菌效力。
基于相容性風險評估至少應該包括以下實驗設計,并進行高溫和光照條件樣品放置。研究包括:原料藥+溶劑;輔料+溶劑;原料藥+輔料+溶劑(關注輔料與原料藥的相互作用)。具體品種可參照《溶液型制劑原輔料相容性研究操作規(guī)程》。
根據(jù)原料藥性質進行以下研究設計:
? 初始注射用水加入量的確定、配液溫度范圍、是否充氮(對氧敏感的單劑量滴眼液)、加料順序、是否避光、pH值調節(jié)范圍、中間體藥液穩(wěn)定性。
? 原輔料若存在結塊現(xiàn)象,影響溶解過程,可考慮增加粉碎過篩工藝,并考察粒徑對溶解時間的影響并做相應研究,粒徑范圍寫入工藝規(guī)程。
根據(jù)溶液的性質進行以下材質研究設計。需提前與廠家確認濾芯材質,濾芯(如PES,PVDF,Nylon,PTFE等)。以過濾效率、含量、濾芯初步相容性為評價指標,初步確定濾芯材質。
藥液的黏度及參比制劑裝量范圍,確定自制品灌裝范圍(結合廠家實際生產設備情況)。
根據(jù)參比制劑包材剖析結果盡量選擇材質一致的包材,材質為塑料瓶+塑料蓋,塑料是半透性材料,會透過氧氣、二氧化碳、水蒸氣等,需關注失水率,同時考察不同來源包材對制劑質量的影響。
制劑滅菌:與注射劑不同的是,滴眼劑大多因為包材的限制(聚乙烯材質),無法采用終端滅菌工藝,而這一點也是可以被認可的。滴眼液的除菌方式一般采用除菌過濾。國外有很多將藥液在配液罐中高溫滅菌后再無菌分裝的操作,但國內不會做此要求。
待小試樣品獲得至少1個月的加速條件穩(wěn)定性和影響因素樣品檢驗結果,且質量變化與參比制劑變化趨勢一致(包括抑菌劑和穩(wěn)定劑含量變化),可開展小試放大,重點關注放大效應。
? 初步穩(wěn)定性考察及確定:包括影響因素試驗(包括低溫、凍融)、加速、中間、長期(具體根據(jù)品種特性確定放樣條件)、開瓶穩(wěn)定性(多劑量包裝)。
? 生產組件相容性及吸附性初步研究: 硅膠管、墊片、不銹鋼等生產組件與溶液進行充分接觸,并設計不同時間點取樣,確認藥液與生產組件的相容性。
? 分析方法初步驗證完成:進行方法確認,時間允許可進行初步方法學驗證,如關鍵耐用性及專屬性試驗,確定質量標準和分析方法SOP。
根據(jù)各國藥典質量研究方法的對比以及小試處方工藝的研究結果,制定最終的標準(原輔料,包材,中間產品,成品),分析方法(中間產品,成品),處方(處方種類,數(shù)量,物料來源)及工藝(詳細的制劑制備工藝)及各個步驟關鍵工藝參數(shù),完成工藝規(guī)程和質量標準備案。
中試放大(GMP)
確定場地及檢測設備無問題,同時與生產場地確認其設備驗證及無菌驗證是否均完成驗證,且是否在有效期內。
包括包材的清洗滅菌、配液參數(shù)、過濾參數(shù)及排液量、灌裝穩(wěn)定性及針頭排液量、中間體存放時限等,關注放大效應。
除關鍵理化性質外,重點關注無菌。
待小試處方確定或中試完成后,可啟動濾芯相容性驗證(可提取物、浸出物分析、化學兼容性、細菌挑戰(zhàn)試驗、潤濕完整性驗證)、包材相容性方法學研究、容器密封性方法學研究(獲得中試樣品后)、生產組件相容性方法學驗證以及抑菌效力驗證。
工藝驗證(GMP)
待小試處方確定或中試完成后,可啟動濾芯相容性驗證(可提取物、浸出物分析、化學兼容性、細菌挑戰(zhàn)試驗、潤濕完整性驗證)、包材相容性方法學研究、容器密封性方法學研究(獲得中試樣品后)、生產組件相容性方法學驗證以及抑菌效力驗證。
2010版中國藥典首次規(guī)定滴眼劑為無菌制劑,《藥品生產質量管理規(guī)范》(2010年修訂)也相應提高了對滴眼劑作為無菌藥品的生產質量管理要求。
無菌藥品的生產工藝一般分為最終滅菌工藝和無菌生產工藝。由于目前滴眼劑瓶材質多為低密度聚乙烯、聚酯類等,在高溫下易變形,因此滴眼劑一般不能采用最終滅菌工藝,需要采用非最終滅菌的無菌生產工藝進行生產。
根據(jù)原料藥性質進行以下研究設計:
? 技術轉移前獲得廠家全部設備驗證及無菌驗證(包括培養(yǎng)基模擬灌裝驗證)報告,且均在有效期內。
? 多劑量滴眼液,技術轉移前需獲得抑菌效力驗證報告。
? 技術轉移批物料到位后,推進廠家提供物料清單,完成確認。
? 獲得驗證批物料檢測報告,完成復核。
根據(jù)原料藥性質進行以下研究設計:
? 關鍵參數(shù)的驗證及中間體存放時限驗證。
? 除菌過濾系統(tǒng)驗證。
? 包材及容器具存放時限驗證(若需和工藝驗證同步完成)。
? 樣品全檢及正式穩(wěn)定性研究。
根據(jù)原料藥性質進行以下研究設計:
待獲得工藝驗證合格樣品后,可進行包材相容性及生產組件相容性、容器密封完整性批樣品檢驗,及安全性評估試驗(包括眼部刺激、過敏試驗等)。
對于真溶液型滴眼劑,因不存在藥物的溶出和釋放過程,美國、歐盟、中國和日本的要求均基本一致,一般要求為:
? 當仿制制劑與參比制劑的濃度規(guī)格一致、輔料種類(Q1)和輔料用量(Q2)一致(用量一致是指仿制制劑與參比制劑的輔料用量差異在±5%范圍內)、物理和化學特性(Q3)對比結果符合要求時,即可豁免體內生物等效性試驗。
? 如果仿制制劑與參比制劑輔料種類不一致或輔料用量與參比制劑差異大于5%、或Q3)對比數(shù)據(jù)不一致,則需進行適當?shù)捏w內生物等效性試驗。
-END-



轉載聲明:未經(jīng)本網(wǎng)或本網(wǎng)權利人授權,不得轉載、摘編或利用其他方式使用上述作品。已經(jīng)本網(wǎng)或本網(wǎng)權利人授權使用作品的,應在授權范圍內使用,并注明“來源:新領先醫(yī)藥科技”。

 010-61006450
010-61006450 聯(lián)系地址:
聯(lián)系地址: 技術市場部:
技術市場部: 010-61006450
010-61006450
